

Descripción
La electroforesis de proteínas de dos dimensiones es una técnica que ha precedido el nacimiento de la proteómica. Y si bien, no es el único esquema utilizado en la proteómica actual, aún es utilizada por sus características y diversas ventajas. Al comienzo de los años 70, la separación proteica de alto rendimiento se realizaba mediante técnicas electroforéticas en presencia de SDS, descrita por Laemmli (Laemmli UK, 1970), una técnica que continúa siendo muy popular, y por otra parte, se realizaba también la separación proteica mediante el isoelectroenfoque, descrito por Gronow y Griffith. No tardó mucho para que estas dos técnicas se unieran, ya que, separaban de manera independiente la masa y el punto isoeléctrico de las proteínas. El primer informe sobre una separación de dos dimensiones (2D) fue en 1974 llevada a cabo por MacGillivray y Rickwood, pero no sorprendieron a la comunidad científica debido a su complejidad y su poca resolución en el gel. No fue sino hasta el siguiente año que O’Farrell mejoró la técnica y mostrando un patrón electroforético mucho mayor debido a que marcó a las proteínas con un isótopo radioactivo de azufre (35S) aumentando la resolución.
Este reporte logró que fuera más fácil la reproducibilidad y la ejecución de la técnica y en consecuencia, permitió la comparación de varios geles en paralelo aumentando la reproducibilidad de las muestras y su comparación. Está técnica es aún usada en nuestros días y poco ha cambiado desde entonces, y aunque el nombre de electroforesis 2D puede sugerir un proceso de dos pasos, en realidad es un proceso de varios pasos y puede durar varios días en su preparación hasta la tinción y se pueden enlistar de manera general de la siguiente manera:
- Extracción de proteínas y preparación de la muestra
- Primera separación por punto isoeléctrico (1D)
- Segunda separación por peso molecular (2D)
- Tinción de los geles (4 días hasta este punto)
- Comparación de cada gel mediante un análisis de imagen
- Finalmente la identificación de las proteínas (western blot, MALDI, LC-MS/MS).
Materiales
- micropipetas
- puntas de 1 mL
- puntas de 20-200 µL
- guantes de nitrilo o latex
- tubos de 15 mL
- tubos de 50 mL
- tubos de 1.5 mL
- gradillas para tubos
- gradilla para geles grandes
- criocajas
- jeringas de 10 mL
- jeringa Hamilton 20 µL
- vaso de precipitados
- parafilm
- pegamento adhesivo de barra
- recipientes de plástico
- matraces de 250 mL
- probetas de diversos volumenes (100 mL – 2 L)
- vortex
- centrifuga
- potenciómetro
- fuente de poder (rango 5000 V y una corriente de 110 mA)
- cámara de 1D
- tubo de vidrio
- embudo de acrilico para geles 1D
- cámara de 2D
- enfriador peltier para cámara de 2D
- cristales para 2D
- cassette para geles de 2D
- Soporte universal
- aro para recipientes
- nuez para soporte universal
- manguera de silicona
- broches para manguera
- acetatos
- neoprenos del tamaño de los cristales de 2D
Reactivos
- Agarosa
- Acrilaimda
- Bis-acrilamida
- Persulfato de amonio
- TEMED
- Tris base
- Tris HCl
- Glicina
- SDS
- Sacarosa
- Polivinilpolipirrolidona (PVPP)
- Urea
- Tiourea
- Tributilfosfato (TBP)
- CHAPS
- Ditiotreitol (DDT)
- Brillant blue G230
- Isopropanol
- Metanol
- Fenol
- Acetato de amonio
- Coomassie
- Anfolitas
- NP-40
Procedimiento
Geles de 1D
- Proceda hacer la extracción proteica como está descrito en el método: gel de acrilamida
- Teniendo las muestras extraídas se almacenan a -80°C hasta su uso
- Prepare alícuotas de acrilamida para los geles de 1D (ver tabla 1)
- Corte los tubos capilares a 23.5 cm de largo (luz 3mm y 1mm de grosor)
- Se coloca en el recipiente del embudo para preparar los geles de 1D con 6 mL de la alícuota de 1D
- Se agregan anfolitas según sean las necesidades del experimento (ejemplo: 108.4 µL anfolitas 3-10 pH, suplementada con 162.1 µL anfolitas 6-8 pH) NOTA: suplementar con otro rango más estrecho de pH enriquece la ventana de entidades electroforéticas, permitiendo mejorar la visualización de las manchas del gel.
- Se agrega a la alícuota 42.5 µL de Persulfato de amonio al 10% y 21 µL de TEMED
- Teniendo la alícuota preparada con las anfolitas y el TEMED se colocan los capilares dentro del embudo para después colocar armar el embudo y el vial (figura 1)
- Se sumerge con mucho cuidado dentro del contenedor para 1D, para que el agua entre por los orificios del embudo y suba por capilaridad la acrilamida en el tubo de vidrio
- Se deja polimerizar 1 hora la acrilamida
- Habiendo polimerizado el gel se procede a colocar cada capilar en la cámara de 1D agregando un empaque en la parte superior
- Se coloca en la parte inferior de la cámara el buffer ácido agregando 1.4 mL de ácido fosfórico al 85% en 2L de agua ultra pura
- Se colocan los capilares junto con la parte interna de la cámara y se agrega el buffer básico agregando 10 mL de NaOH 10N y 990 mL de agua ultra pura. NOTA: tener cuidado de no mezclar o salpicar por ningun motivo ambos buffer o de que no haya fugas, ya que esto ocasionara que no se de un enfoque de proteínas efectivo.
- Teniendo ambos buffers colocados retire las burbujas de aire dentro de los capilares agregando el buffer básico con una jeriga hamilton
- Coloque posteriormente de 30-50 µL de buffer de solubilización en una dilución 2:1. NOTA: el buffer puede estar teñido con azul de bromofenol para mejorar su visualización. Este paso es importante ya que se evita el contacto de la muestra proteica con el NaOH
- Cierre la cámara y conecte a la fuente de poder y proceda a realizar un pre-enfoque a 1000 V por 45 min.
- Después quite la tapa y cargue con una jeringa hamilton 500 µg de proteína extraidas y solubilizadas como esta descrito en método: gel de acrilamida
- Cargada la muestra cierre la cámara y conectela a la fuente de poder a 1000 V/h durante 23.5 horas. NOTA: coloque alguna marca en el tubo para no perder el orden de las muestras.
- Antes de terminar la corrida usted debe preparar los geles para 2D con 2 horas de anticipación (ver abajo). NOTA: si usted carece de experiencia en geles 2D tome sus precauciones y aumente el tiempo
- Terminada la corrida de los geles de 1D retire el buffer básico de los capilares y coloque los capilares sobre hielo
- Utilice una jeringa y un expulsor de geles como se muestra en la figura 2
- Retirado el gel, se coloca en un contenedor con buffer de equilibrio (ver tabla 2) y se deja incubando por 5 min cambie el buffer dos veces y deje otros 5 min. NOTA: existen métodos rápidos para visualizar las bandas de la 1D sin afectar la 2D.
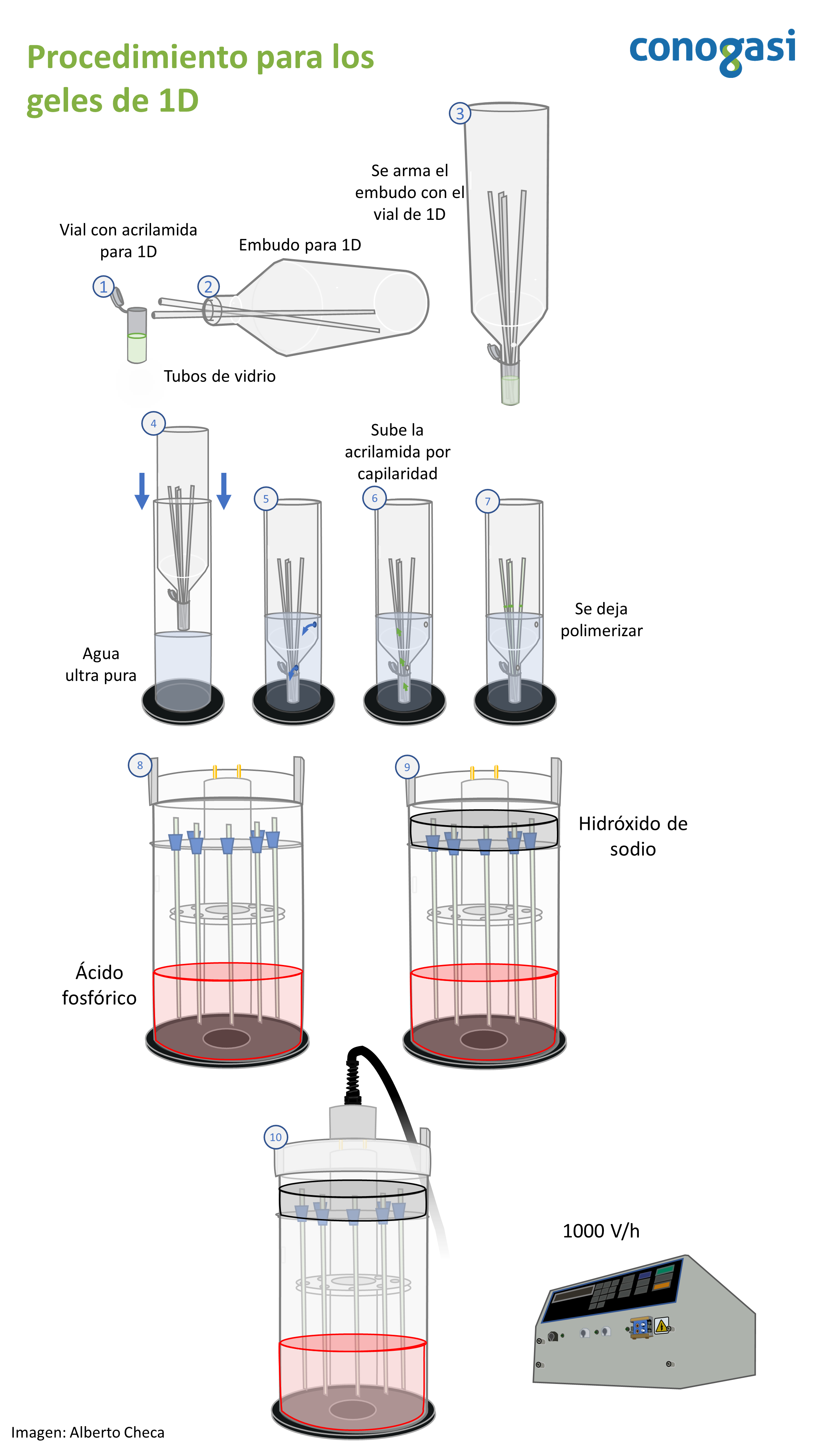
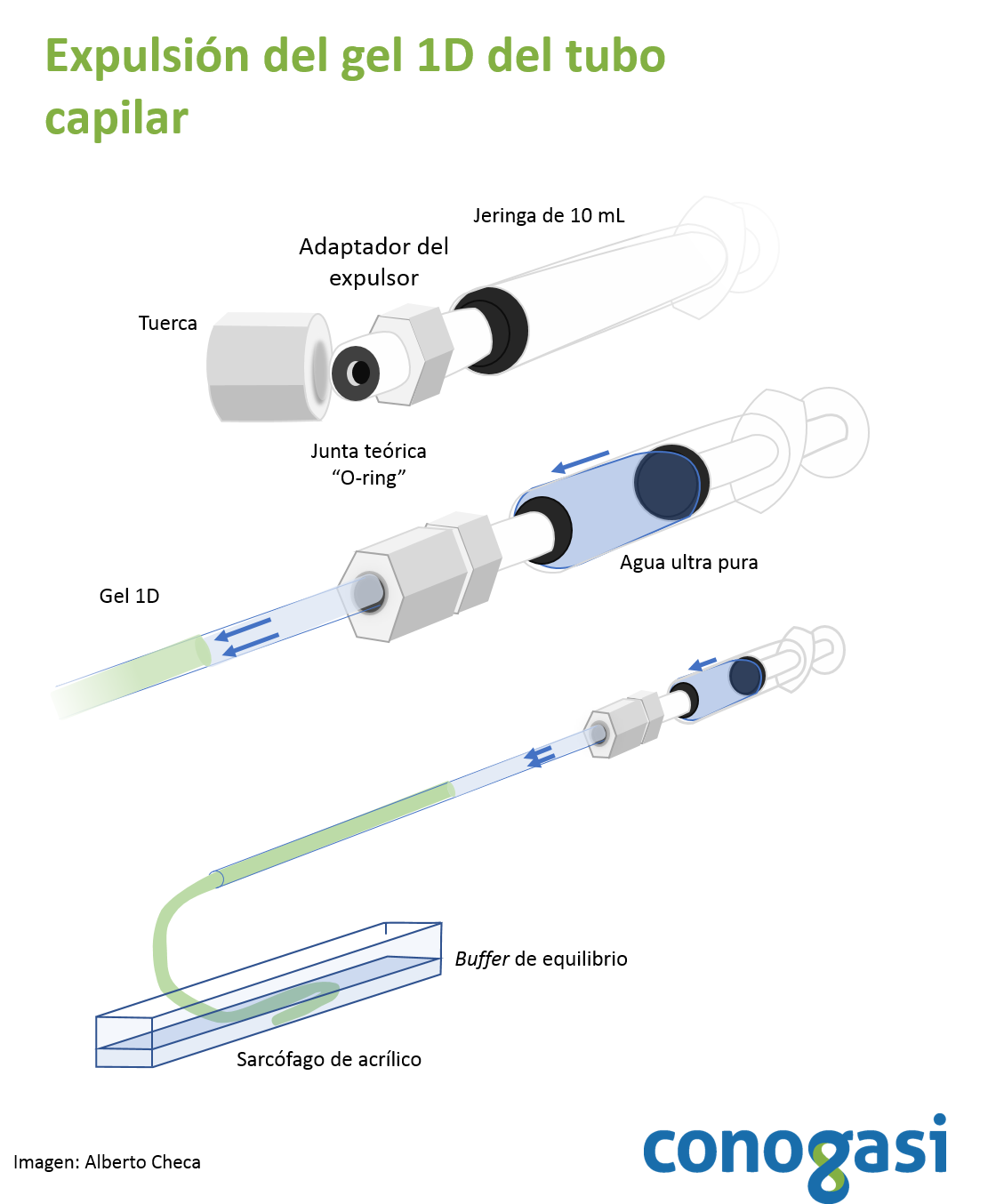
Tabla 1. Alícuotas de 1D
| Urea 9.5M | 57 g |
| NP-40 | 2 mL |
| CHAPS 5M | 0.3 g |
| Acril/bis-acril 30:8 (p/p) | 13 mL |
| Agua ultra pura | 95 mL |
NOTA: las alícuotas se filtran con una membrana de 0.45 µm. La urea se debe disolver en baño maría a 37°C evitando que suba la temperatura
Tabla 2. Buffer de equilibrio
NOTA: Se prepara la disolución A y se almacena. La disolución B se prepara hasta que se va utilizar agregando
el DTT y el azul de bromofenol en 100 mL de disol. A.
| A | Tris Base 8 mM | 1.06 g |
| Tris HCl 300mM | 47.8 g | |
| SDS 3% | 30.0 g | |
| Aforar a | 1000 mL | |
| B | Azul de bromofenol | 100 µg |
| DTT | 770 mg | |
| Aforar con a | 100 mL |
Geles 2D
NOTA: Antes de que finalice la 1D usted debe preparar sus geles para la segunda dimensión
- Antes de armar su caster gel verifique que todo su material esta previamente lavado y seco. NOTA: use mezcla crómica para lavar el material de vidrio
- Arme el caster gel de 2D como se indica en la figura 3
- Prepare el soporte universal con el vaso que contendrá la acrilamida para los geles de 2D
- Prepare la disolución de acrilamida para los geles en un vaso de precipitados y sobre un agitador magnético (Tabla 3). NOTA: puede utilizar un clip en lugar de un magneto de mosca.
- Teniendo la mezcla de los geles se coloca en el recipiente unido al caster gel evitando en todo momento hacer burbujas. NOTA: Generalmente se preparan los geles al 12%
- Abra las pinzas de presión con mucho cuidado para que la acrilamida entre al caster y evitando la formación de burbujas. NOTA: si usted observa burbujas puede dar unos golpes sutiles al caster
- La acrilamida debe llegar hasta el borde de los vidrios evitando derrames
- Posteriormente agrega isopropanol al 50% y espere aprox. 1h o hasta alcanzar totalmente la polimerización observando el remanente en el vaso unido al caster gel. NOTA: varios factores como la temperatura y el oxígeno pueden afectar la polimerización, tome sus precauciones.
- Habiendo terminada la polimerización desarme con mucho cuidado el caster gel
- Lave con agua corriente los vidrios quitando los remanentes de la polimerización
- Coloque los vidrios previamente lavados en una gradilla
- Coloque las tripas de los geles de 1D y adhiéralas con agarosa al 1% y Tris-Gly-SDS 1X. NOTA: agregue azul de bromofenol en la agarosa para visualizar el frente de corrida.
- Coloque los vidrios en la cámara de 2D.
- Llene el tanque de la cámara con Tris-Gly-SDS 1X en la parte de abajo y coloque los empaques si su cámara los lleva.
- Llene hasta donde indique su cámara en la parte superior con Tris-Gly-SDS 2X.
- Cierre la cámara y programe su fuente de poder a 1000V/h durante 24 horas. NOTA: usted puede aumentar el voltaje y correr sus geles en menos tiempo pero esto requiere de optimización.
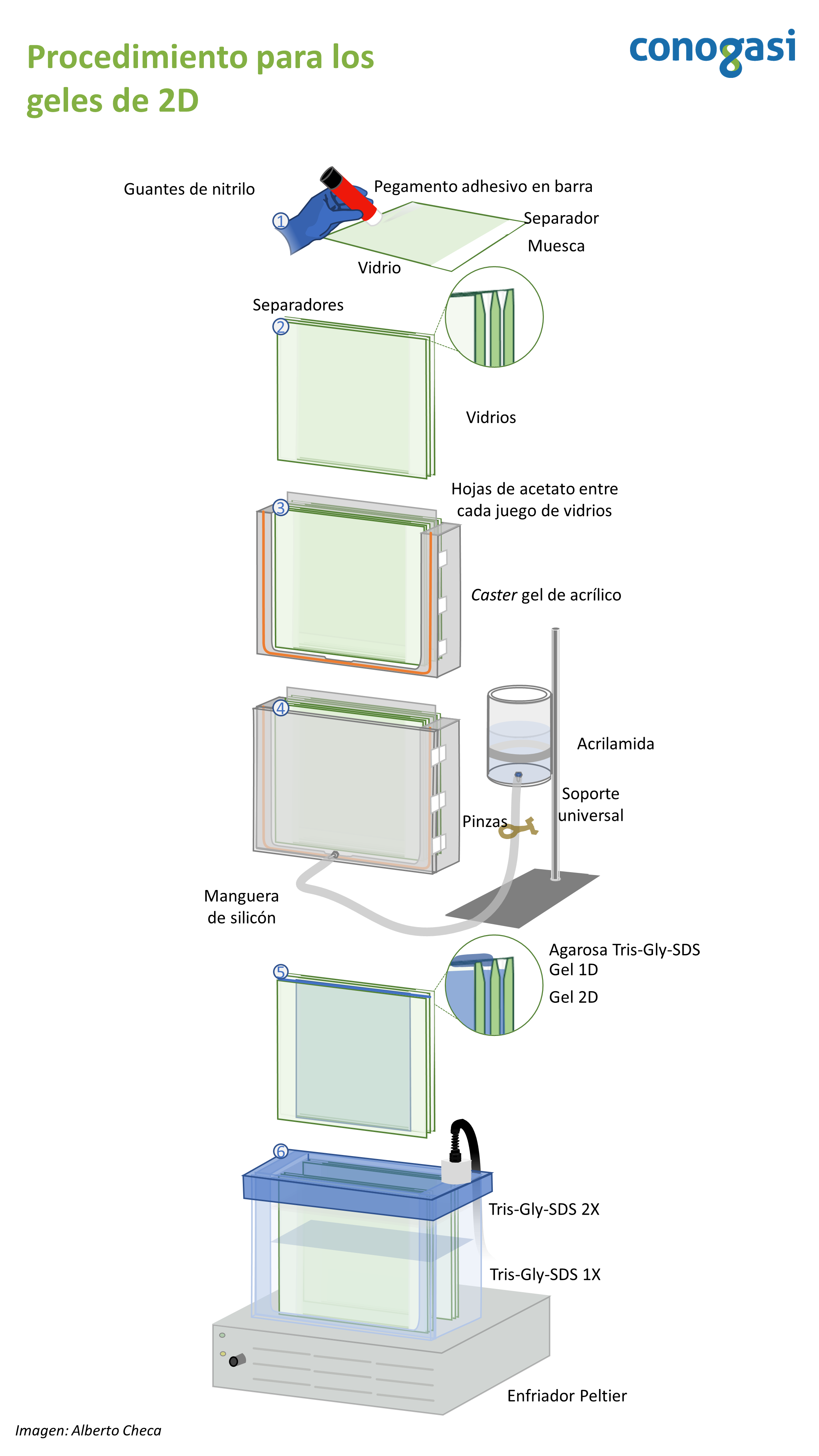
Tabla 3. Geles de 2D
| Formato grande | 6 geles (12%) |
| Acrilamida/bis 30% | 211.0 mL |
| Tri HCl pH 8.8 | 130.5 mL |
| Agua ultra pura | 180.5 mL |
| SDS 10% | 11.70 mL |
| Persulfato de Amonio 10% | 2.900 mL |
| TEMED | 0.376 mL |
| Volumen Final | 537.0 mL |
Resultado

Cómo citar: Checa Rojas, A., Mitzy Ríos de Anda (2018, 16 de Enero ) Método: Gel de poliacrilamida de dos dimensiones 2D-PAGE. Conogasi, Conocimiento para la vida. Fecha de consulta: Septiembre 18, 2025
Esta obra está disponible bajo una licencia de Creative Commons Reconocimiento-No Comercial Compartir Igual 4.0
Deja un comentario
Sé el primero en comentar!