

Introducción
La electroforesis es un método por el cual se separan moléculas por tamaño o peso molecular usando un campo eléctrico. La separación se realiza comúnmente en un gel que funciona como filtro poroso y está constituido habitualmente de materiales como agarosa o poliacrilamida. Su principal uso es la separación de mezclas de biopolímeros especialmente de ácidos nucleicos (ADN o ARN) o proteínas. Los ácidos nucleicos disponen de una carga eléctrica negativa y cuando son separados en el gel mediante una corriente eléctrica, se dirigirán al polo positivo. A diferencia de las proteínas que, se cargan mediante una molécula llamada SDS (dodecilsulfato de sodio) que se incorpora a las proteínas, adquiriendo una carga negativa. Al colocar una muestra de proteínas dentro del gel y someterlas a una carga eléctrica, éstas migrarán y se moverán a través del gel hacia el polo positivo. Las moléculas más pequeñas se moverán más rápidamente y las más grandes quedarán cerca del lugar de partida en el gel.
La aplicación de la electroforesis en la investigación biomédica es muy diversa, desde cuestiones muy básicas como analizar la calidad de una extracción de ácidos nucleicos o proteínas o como un paso previo para la purificación u otros métodos empleados en biología molecular o bioquímica para el análisis de muestras de pacientes para el diagnóstico de enfermedades, análisis de ascendencia, reconocimiento de cadáveres, pruebas de paternidad, etc.
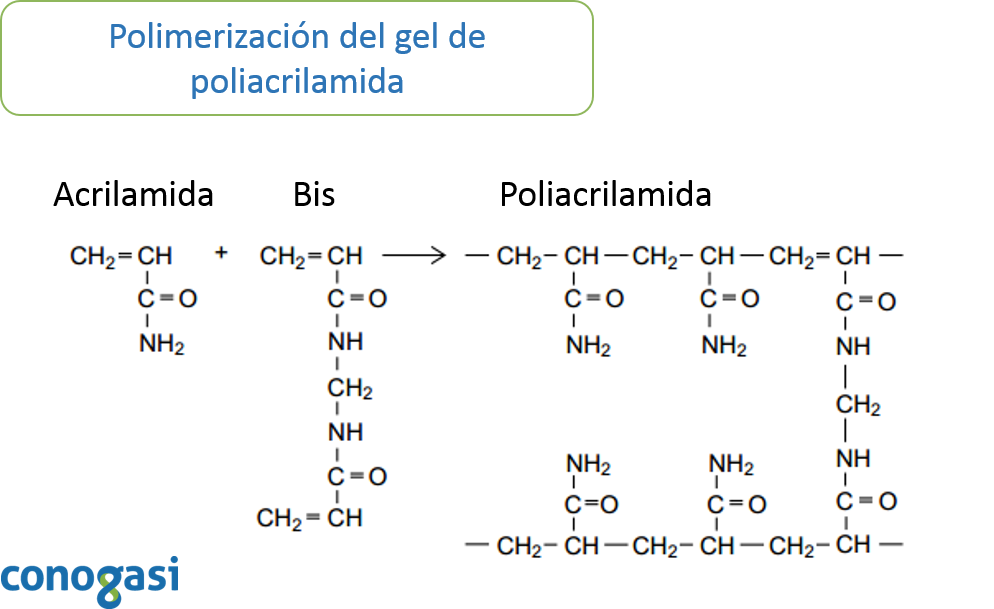
Para el análisis de proteínas, la electroforesis en gel puede proporcionar información sobre el peso molecular, la carga, las subunidades o la pureza de una preparación. Es relativamente simple de usar y es altamente reproducible. El uso más común es el análisis cualitativo de mezclas complejas de proteínas. Los geles de poliacrilamida se forman por la copolimerización de la acrilamida y la bis-acrilamida. La reacción es una adición de polimerización vinílica iniciada por la generación de radicales libres (Chrambach, 1985). La polimerización inicia cuando se agregan el persulfato de amonio y el TEMED, el cual, acelera la tasa de formación de radicales libres del persulfato que a su vez catalizan la polimerización. Los radicales libres del persulfato convierten los monómeros de acrilamida en radicales libres los cuales reaccionan con los monómeros inactivados para empezar la reacción en cadena de la polimerización (Shi and Jackowski, 1998). La elongación de la cadena del polímero se entrelaza aleatoriamente con la bis-acrilamida, resultando en un gel con la porosidad deseada la cual depende de la concentración de los monómeros.
Materiales
- Muestra biológica
- Mortero de cerámica
- Guantes
- Micropipetas
- Gradillas
- Tubos de 1.5 mL
- Tubos de 15 mL
- Tubos de 50 mL
- Centrífuga
- Cámara de electroforesis
- Fuente de poder
- Escáner
- Congelador
Reactivos
- Nitrógeno líquido
- Inhibidores de proteasas y fosfatasas
- Fenol
- Acetona
- Metanol
- Acetato de amonio.
- acrilamida
- bis-acrilamida
- SDS
- Persulfato de amonio
- Sulfato de amonio
- 2-mercaptoetanol
- Tris HCl
- Tris Base
- Coomassie
- Ácido acético
- KCl
- PVPP
- HCl
- EDTA
- Sacarosa
- Urea
- Tiourea
- Chaps
- TBP
- DTT
- Temed
Procedimiento
1. Extracción fenólica de proteínas de tejido, células en cultivo: suspensión o adherentes.
NOTA: existen diversos métodos de extracción de proteínas. Este método es compatible para geles de 1D y 2D-SDS PAGE
- Cuando se utiliza tejido es necesario congelarlo y después macerarlo en nitrógeno líquido sobre un mortero de cerámica o vidrio. En caso de usar células en cultivo pasar al paso 3.
- Se macera el tejido en un mortero evitando que se descongele y se vea pastoso el tejido. Se presiona la muestra con el pistilo hasta que se observe un polvo muy fino.
- Retirar el medio por centrifugación a 2,000 x g durante 4 minutos. Una vez obtenido el botón celular o el polvo de tejido macerado se transfiera a un tubo 1.5 mL con 1 mL de una disolución acuosa con inhibidores de proteasas y fosfatasas.
- Se coloca la muestra en un sonicador y se dan 3 pulsos de 1-5 segundos dejando reposar la muestra en intervalos de 10 segundos sobre hielo.
- Después, se agregan 5 volúmenes de acetona al 99.99% y se deja 1-2 horas a menos 20 grados Celsius (-20°C).
- Posteriormente se centrifuga a 5,000 x g durante 10 min y se retira el sobrenadante, dejando evaporar la acetona del botón.
- Se agrega 500 µL de buffer de extracción (ver tabla 1) y 600 µL de fenol sobre el botón, se le da vortex durante 10 min.
- Se centrifuga a 4,000 x g por 10 min y se recupera la parte acuosa (superior).
- Se agregan 3 volúmenes de acetato de amonio 0.1 M diluido en metanol y se deja precipitando ≥1 h a menos 70 grados Celsius (-70°C).
- Se centrifuga a 4,000 x g por 20 min y se lava 3 veces con acetato de amonio-metanol. El último lavado se realiza con acetona al 99.9%
- Se evapora la acetona en una centrífuga de vacío (speed vac) o se deja evaporar boca abajo. NOTA: no lleve a sequedad total.
- Se solubiliza el botón con el buffer de solubilización (ver tabla 2) con 50-500 µL dependiendo del tamaño de la muestra. Este buffer de solubilización es compatible para geles 2D-PAGE (para más detalles consultar el método de Geles de 2D-PAGE)
- Centrifugar a 13,000 x g por 20 min para retirar el material insoluble, deposite el sobrenadante en un tubo nuevo y cuantifique la muestra.
Tabla 1. Buffer de extracción
| Concentración Final | Reactivo | para 200 mL |
| 0.7 M | Sacarosa | 47.922 g |
| 0.5 M | Tris-Base | 12.11 g |
| 0.1 M | KCl | 1.4 g |
| 30 mM | HCl | 0.48 mL |
| 50 mM | EDTA (292.24 g/mol) | 2.9224 g |
| 2% v/v | 2-Mercaptoetanol | 4 mL |
| 1.2% p/v | PVPP | 2.4 g |
Tabla 2. Buffer de solubilización de proteínas
| Concentración Final | Reactivo | Para 5mL |
| 7 M | Urea | 2.1033 g |
| 2 M | Tiourea | 0.761 g |
| 4% | CHAPS | 0.2 g |
| 2 mM | TBP | 0.05 mL |
| 2% | Anfolinas 3-10pH | 0.1 mL |
| 60 mM | DTT | 0.04627 g |
Cuantificación de proteínas por el método de Bradford ácido.
- Se solubiliza 100 mg de albúmina bovina en 1 mL y realizan diluciones seriadas desde 10 ng/µL hasta 1 mg/µL para realizar la curva estándar.
- Se agregan 4 µL de HCl 0.12N y 36 µL de muestra de proteínas e incubar por 5 minutos a temperatura ambiente.
- Se agrega 1.4 mL de disolución concentrada de Bradford a cada muestra (Diluir Bradford Dye, Bio-Rad) en agua ultra pura a una proporción de 1:3 y filtrar con papel Wattman No1).
- Después se agrega la disolución de Bradford diluida a las muestras ácidas y se aplica agitación evitando burbujas.
- Se coloca el Bradford a cada muestra y se coloca además un tubo el cual contiene todo lo anterior menos proteínas en su lugar se agrega solo el buffer de solubilización (blanco).
- Se colocan las muestras y el blanco en cada celda o en placas para espectrofotometría
- Se colocan las celdas o la placa dentro del espectrofotómetro y se cuantifica a una absorbancia a 595 nm.
- Se grafican los valores de absorbancia de la curva estándar y se realiza una interpolación de los valores de las muestras problemas para estimar la concentración de proteína de cada muestra.
Polimerización del gel de poliacrilamida
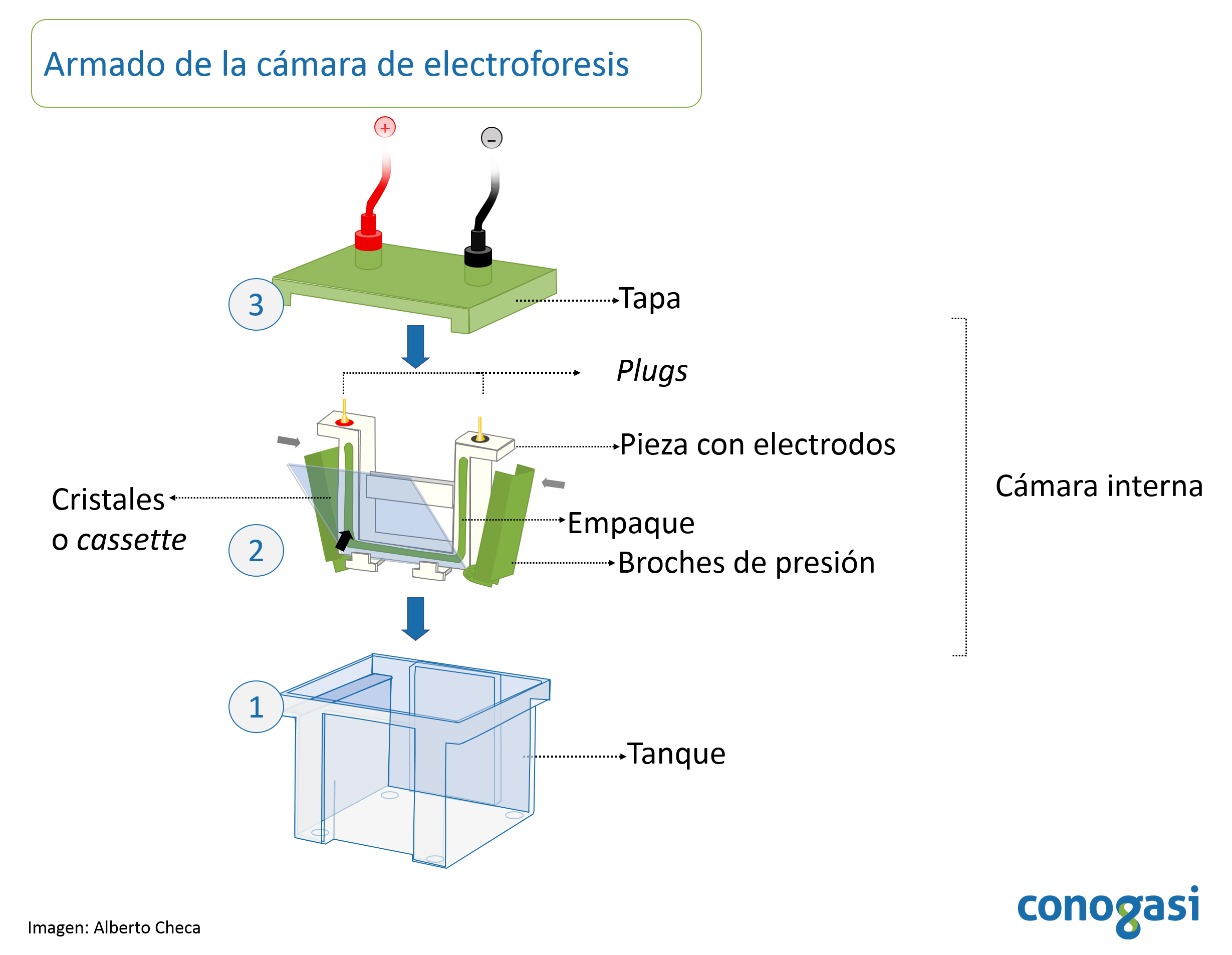
- Se arma la camara y se colocan los vidrios según las características del proveedor de la cámara de electroforesis (figura 1).
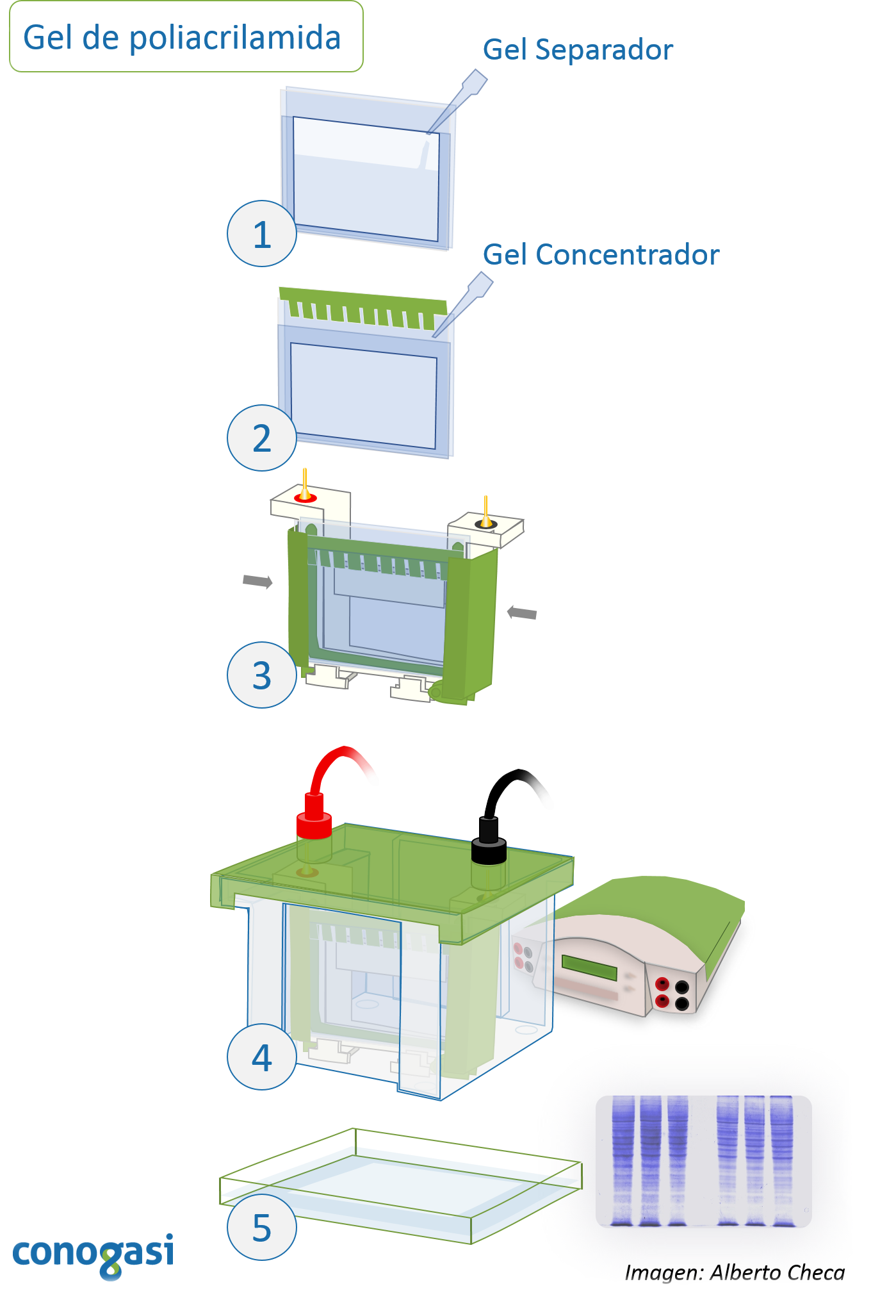
- Se prepara la disolución de acrilamida separador a la concentración deseada (ver tabla 4) y la disolución de acrilamida concentrador en dos tubos de 15 mL (ver tabla siguiente)
- Se coloca en un tubo de 1.5 mL, un mililitro de la mezcla de acrilamida separador con persulfato de amonio y agrega 0.5 µL de TEMED y se resuspende rápidamente sin hacer burbujas.
- Se deposita esta mezcla sobre un extremo pegado al vidrio de tal modo que se hará un tapón en la parte inferior (esto evitará que se salga la mezcla).
- Se deja polimerizar el tapón y se cerciora de que el tapón funciona depositando agua ultra pura con una piseta. Se retira el agua y se retiran todas las gotas.
- Posteriormente se coloca la disolución separadora de acrilamida con 8.5 µL de TEMED y se resuspende rápidamente.
- Se coloca cuidadosamente sobre un extremo de los vidrios sin llegar al borde dejando espacio suficiente para colocar la mezcla de acrilamida concentradora. Luego de haber colocado esta disolución se coloca alrededor de 20-50 µL de isopropanol al 50% para romper las burbujas que se hayan formado y aplanar el gel.
- Se deja polimerizar el gel. NOTA: se debe retira el exceso de isopropanol cuando esté polimerizado el gel.
- Terminando la polimerización (20 – 30 min aprox.) se coloca la disolución de acrilamida concentradora con 2.5 µL de TEMED y se resuspende la disolución
- Se coloca la mezcla encima del gel polimerizado con mucho cuidado se coloca el peine evitando la formación de burbujas. Se deja polimerizar el gel y se deja hasta que estén listas las muestras de proteínas.
- Algunos equipos deben colocarse un adaptador a los vidrios para después ser colocados dentro de la cámara (ver instructivo de la cámara del proveedor).
Disoluciones para los geles de poliacrilamida
| Stock Acrilamida-bis 30% | 1 L | 500 mL |
| Acrilamida | 300 g | 150 g |
| Bis-acrilamida | 8 g | 4 g |
Disolución Buffer: Tris pH 8.8; 1.5M y Tris pH 6.8; 0.5M
| Tris pH 8.8, 1.5 M | 1L | 500 mL |
| Tris-base | 130.8 g | 65.4 g |
| Tris-HCl | 66.3 g | 33.15 g |
| Tris pH 6.8, 0.5M | 1L | 500 mL |
| Tris-base | 1.71 g | 0.855 g |
| Tris-HCl | 76.8 g | 38.40 g |
Tabla 3. Buffer de Carga para mini-geles de acrilamida
| 2X (Volumen) | Reactivo |
| 0.8 mL | 1,5M de Tris (6.8) |
| 4.0 mL | SDS 10% |
| 1.0 mL | β-Mercaptoetanol |
| 2.0 mL | Glicerol 100% (p/V) |
| 20-200 µg | Azul de Bromofenol B.B. |
| 2.0 mL | H2O |
| 10.0 mL | Vol. Final |
NOTA: la cantidad de azul de bromofenol dependera de que tan intensa se quiera ver el colorante en el buffer para su visualización en el gel cuando la muestra este corriendo.
Tabla 4. Reactivos para minigeles de poliacrilamida
| Separador | |||||
| Reactivos | 10% | 12% | 13.5% | 15% | 18% |
| Acrilamida 30% (mL) | 3.33 | 4.0 | 4.5 | 5.0 | 6.0 |
| Tris pH 8.8 (mL) | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 |
| SDS 10% (mL) | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| H20 ultra pura (mL) | 4.0 | 3.33 | 2.83 | 2.33 | 1.33 |
| Persulfato de amonio 10% (µL) | 40 | 40 | 40 | 40 | 40 |
| TEMED (µL) | 8.5 | 8.5 | 8.5 | 8.5 | 8.5 |
| Concentrador para más de 2 geles | |
| Acrilamida-bis 30% | 660 µL |
| Tris pH 6.8, 0.5M | 1.25 mL |
| SDS 10% | 50 µL |
| H20 ultrapura | 2.97 mL |
| Persulfato de amonio 10% | 50 µL |
| TEMED | 2.5 µL |
Nota: antes de agregar el Persulfato y el temed se pueden hacer alicuotas y almacenar los tubos protegidos de la luz.
Carga de las muestras en el gel y corrida
Una vez teniendo cuantificada la muestra se mezcla con un buffer de carga (ver tabla 3) y se coloca dentro del gel en un orificio diseñado para depositar la muestra.
- En el primer pozo u orificio se colocan marcadores de pesos moleculares que ayudaran a saber cuál es el peso molecular de nuestras bandas de interés observadas en el gel (por lo general estos ya contienen buffer de carga. NOTA: ver características técnicas en la ficha de su proveedor).
- En los siguientes pozos u orificios se colocará las muestras en una concentración entre 10-50 µg contenido en un volumen máximo de 5-10 µL de muestra por pozo.
- Se coloca buffer de corrida 1X dentro en la parte donde va nuestro gel hasta que toque el gel
- Se coloca buffer de corrida en la cámara de electroforesis hasta la mitad del gel.
- Se tapa cuidadosamente la cámara y se conecta a la fuente de poder a 45mA (dos geles) o 120V por 45 min a 1hora. NOTA: se puede modificar el voltaje según las necesidades del usuario
- Se debe detener la corrida del gel cuando el azul de bromofenol llega a la parte inferior del gel.
- Se desconecta de la fuente de poder y se retiran los vidrios que contienen el gel y se saca con mucho cuidado. NOTA: si el gel será utilizado en western blot hasta aquí termina este procedimiento.
- Se coloca el gel en un recipiente con una disolución de metanol-ác. acético (4:1).
- Se deja 5 min y se lava abundantemente con agua ultra pura o bidestilada, hasta dejar de percibir el acético.
- En un recipiente limpio se coloca 30-50 mL de disolución de Coomassie y se deja tiñendo en agitación ≥2 horas.
- Posterior a la tinción se retira el gel y se coloca en un recipiente limpio y se lava con agua ultrapura o bidestilada hasta quitar el exceso de colorante.
- Se coloca en un escáner y se guarda la imagen para visualizarla digitalmente.
Preparación del Coomassie coloidal
- Disolver 200g de sulfato de amonio ((NH4)2SO4 ) en 400 mL de agua ultra pura en una placa de agitación. Posteriormente filtrar con una membrana de PVDF de 0.45µm
- Agregar 200mL de Ácido Fosfórico en agitación dejar 1min y posteriormente agregar 2.4 g de Coomassie Brillant blue G-250
- Agitar hasta disolver el Coomassie y agregar 880mL de agua ultra pura o bidestilada hasta homogenizar
- Posteriormente agregar 400mL de metanol 99%
- El coomassie debe verse azul-verdoso de ser así almacenar en una botella ámbar hasta su uso.
Reactivos para la preparación del Coomassie
| Volumen de la solución | |||
| Para preparar: | 500mL | 1L | 1.5 L |
| Sulfato de amonio (NH4)2SO4 | 50 g | 100 g | 150 g |
| Agua | 100 mL | 200 mL | 300 mL |
| H3PO4 85% | 50 mL | 100 mL | 150 mL |
| Coomassie | 0.6g | 1.2 g | 1.8 g |
| Agua ultra pura | 220 mL | 440 mL | 660 mL |
| Etanol CH3CH2OH | 100 mL | 200 mL | 300 mL |
Disolución de fijado
| Fijar | 500 mL | 1 L | 1.5 L | 2 L | Tiempo |
| 50% de met-OH | 250 mL | 500 mL | 750 mL | 1000 mL | Al menos 30min en agitación |
| 10% Ac. Acético | 50 mL | 100 mL | 150 mL | 200 mL | |
| 40% agua ultra pura | 200 mL | 400 mL | 600 mL | 800mL | |
| Tinción Coomassie col. | Al menos 1hr agitación | ||||
| Disolución para Desteñir | 500 mL | 1 L | 1.5 L | 2 L | Al menos 30min en agitación |
| 5% met-OH | 25 mL | 50 mL | 75 mL | 100 mL | |
| 7% Ac. Acético | 35 mL | 70 mL | 105 mL | 140 mL | |
| agua ultra pura | 440 mL | 880 mL | 1320 mL | 1760 mL | |
Buffer Tris glicina SDS, Tris-Gly-SDS o Buffer de Corrida
| Reactivos | 1X | 5X | 10X |
| Glicina | 14.4 g | 72 g | 144.0 g |
| Tris Base | 3.0 g | 15 g | 30 g |
| SDS | 1 g | 5 g | 10 g |
| H2O ulta pura o bidestilada | 1000 mL | 1000 mL | 1000 mL |
Resultados
Una mezcla de proteínas que serpa separada por peso molecular mediante el gel de poliacrilamida. Las moléculas separadas lucen como múltiples bandas, distribuidas a lo largo del gel.
Desechos
Los geles de poliacrilamida se utilizan comúnmente en la investigación en bioquímica y biología molecular. Aunque la acrilamida polimerizada no está regulada como residuo peligroso, los geles de poliacrilamida pueden contienen acrilamida residual sin polimerizar, que es un material tóxico que puede representar un peligro cuando se introduce en el medio ambiente. Utilice las siguientes pautas al desechar geles de poliacrilamida
1. Los geles de poliacrilamida deben eliminarse a través de un Programa de Desechos Químicos de su área de trabajo. No deseche los geles de poliacrilamida en la basura normal (muncipal) o en bolsas rojas como desechos biológicos.
2. Coloquelos los geles dentro de una bolsa a prueba de fugas. Selle la bolsa y coloque la bolsa sellada dentro de una caja de cartón bien etiquetada
3. La etiqueta debe de tener un color llamativo e identificado como “RESIDUOS QUÍMICOS” e identificado como desechos de “geles de poliacrilamida”
4. Una vez llena la caja esta debe de ser transportada por una compañia especializada en manejo de desechos químicos o a través del Programa de Desechos Químicos de su institución o empresa.
5. Los guantes y la suciedad visiblemente contaminados con geles de poliacrilamida deben colocarse en una bolsa de plástico sellada separada. Al igual que los geles puede colocarla la bolsa sellada dentro de una caja de cartón y etiquetada como se indicó en la parte de arriba. Elimine a través del Programa de Desechos Químicos.
Notas
La muestra embebida en un gel puede observarse a simple vista en el caso de una tinción colorida (en proteínas, ver Método tinción Coomassie), o puede ser teñida con colorantes fluorescentes (ver Métodos de tinción Fluorescente para proteínas).
Fuentes de error
Todos los materiales y reactivos que se usan deberán estar químicamente limpios y estériles. Además, deben estar libres de proteasas con el fin de evitar la degradación de la muestra.
APLICACIONES
Coinmunopresipitación
Análisis de interacción de proteínas
Proteómica: geles 1D
Espectrometría de masas: MALDI-TOF y LC-MS/MS
Cómo citar: Checa Rojas, A. (2017, 26 de Junio ) Método: Gel de poliacrilamida para proteínas. Conogasi, Conocimiento para la vida. Fecha de consulta: Julio 18, 2025
Esta obra está disponible bajo una licencia de Creative Commons Reconocimiento-No Comercial Compartir Igual 4.0
Deja un comentario
2 Comentarios en "Método: Gel de poliacrilamida para proteínas"
Muy buena la información, pero hay tablas en las que no se especifican las unidades
¿Cómo se desecha el gel una vez terminada la práctica? ¿Cuál es su correcta gestión?